BilayerGrapheneGenerator¶
-
class
sknano.generators.BilayerGrapheneGenerator(*args, autogen=True, finalize=True, **kwargs)[source] [edit on github][source]¶ Bases:
sknano.generators.GrapheneGenerator,sknano.core.structures.BilayerGrapheneBilayer graphene structure generator class.
Parameters: - armchair_edge_length (float, optional) –
Length of armchair edge in Angstroms
New in version 0.3.10.
- zigzag_edge_length (float, optional) –
Length of zigzag edge in Angstroms
New in version 0.3.10.
- length (float, optional) –
Length of armchair edge in Angstroms
Deprecated since version 0.3.10: Use
armchair_edge_lengthinstead - width (float, optional) –
Width of graphene sheet in Angstroms
Deprecated since version 0.3.10: Use
zigzag_edge_lengthinstead - edge ({'AC', 'armchair', 'ZZ', 'zigzag'}, optional) –
ArmChair or ZigZag edge along the
lengthof the sheet.Deprecated since version 0.3.10: No longer used!
- basis ({
list}, optional) –List of
strs of element symbols or atomic number of the two atom basis (default: [‘C’, ‘C’])New in version 0.3.10.
- element2 (element1,) –
Element symbol or atomic number of basis
Atom1 and 2Deprecated since version 0.3.10: Use
basisinstead - bond (float, optional) – bond length between nearest-neighbor atoms in Angstroms.
- layer_spacing (float, optional) – Distance between layers in Angstroms.
- stacking_order ({'AA', 'AB'}, optional) – Stacking order of graphene layers
- layer_rotation_angle ({None, float}, optional) – Rotation angle of second layer specified in degrees.
If specified in degrees, then you must set
degrees=True - degrees (bool, optional) – The
layer_rotation_angleis specified in degrees and needs to be converted to radians. - autogen (bool, optional) – if
True, automatically generate unit cell and full structure - verbose (bool, optional) – verbose output
Examples
Import the BilayerGrapheneGenerator class
>>> from sknano.generators import BilayerGrapheneGenerator
Generate 100 Å wide by 10 Å long
ABstacked bilayer-graphene:>>> bilayer_graphene = BilayerGrapheneGenerator(armchair_edge_length=100, ... zigzag_edge_length=10)
Save structure data in
xyzformat:>>> bilayer_graphene.save()
Here’s the rendered structure (after rotating 90 degrees so that it better fits the page):

The next command generates bilayer-graphene with its second layer rotated 45 degrees.
>>> rotated_bilayer = ... BilayerGrapheneGenerator(armchair_edge_length=50, ... zigzag_edge_length=50, ... layer_rotation_angles=[0, 45], ... degrees=True) >>> rotated_bilayer.save(fname='rotated_bilayer.xyz')
A rendering of
rotated_bilayer: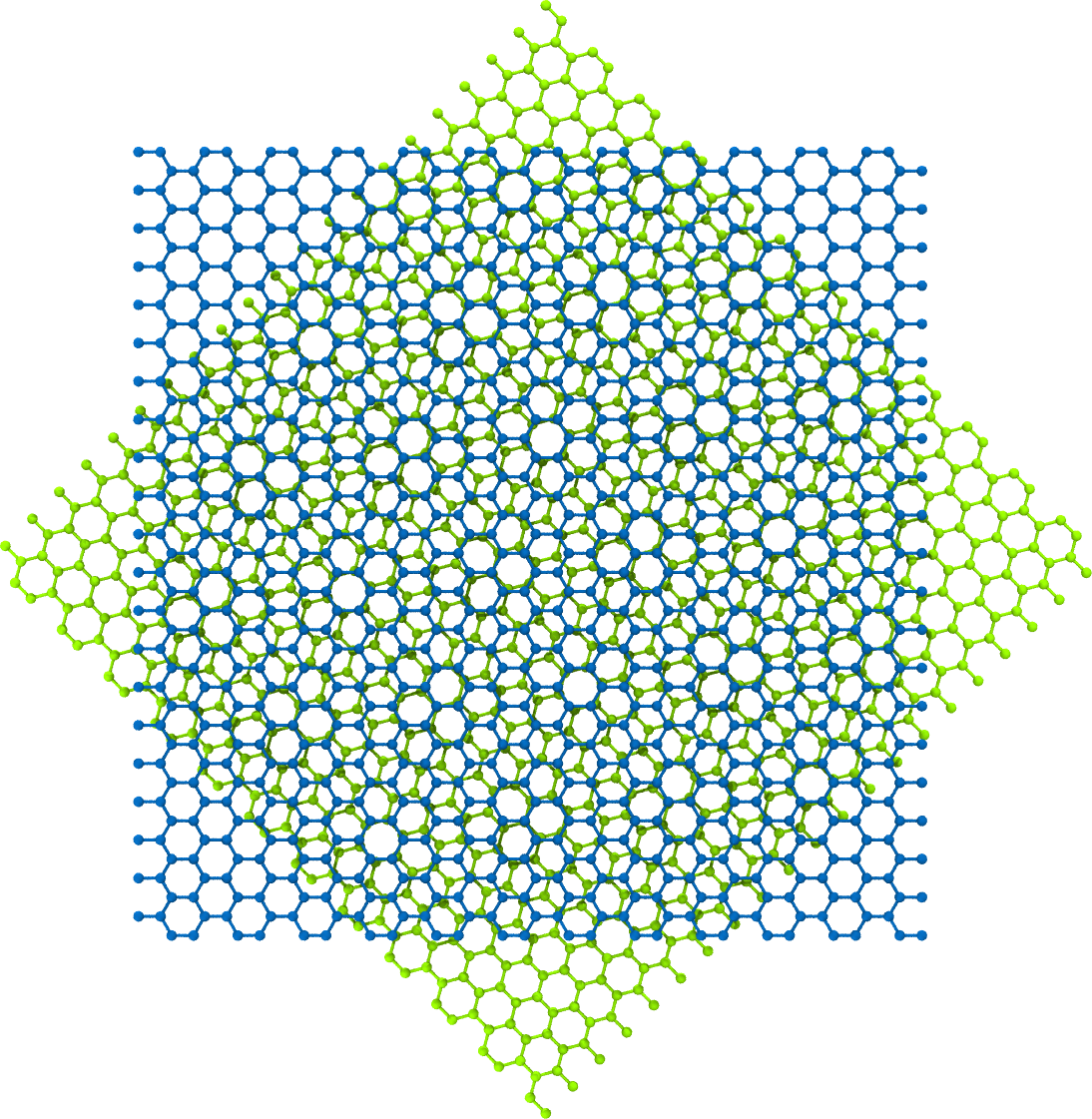
The next example is identical to the one above, except the atomic basis is Boron and Nitrogen.
>>> rotated_BN_bilayer = ... BilayerGrapheneGenerator(armchair_edge_length=50, ... zigzag_edge_length=50, ... basis=['B', 'N'], ... layer_rotation_angles=[0, 45], ... degrees=True) >>> rotated_BN_bilayer.save(fname='rotated_bilayer_B-N_basis.xyz')
The rendered structure looks like:
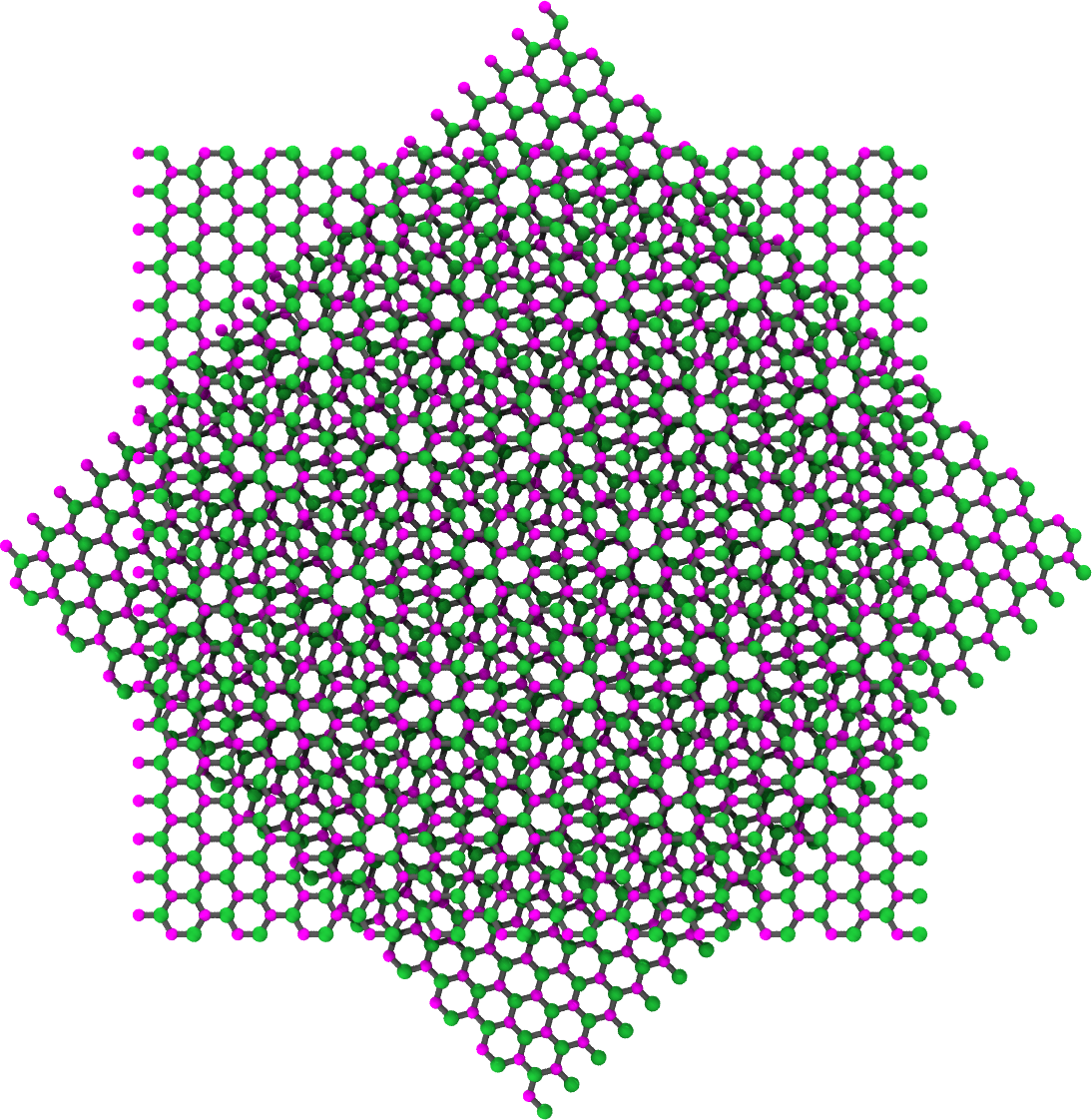
Attributes
NNumber of graphene unit cells. NatomsN atoms. Natoms_per_layerNumber of atoms per layer. Natoms_per_unit_cellNumber of atoms per unit cell. areaTotal area of graphene supercell. atomsStructure Atoms.basisNanoStructureBasebasis objects.crystal_cellStructure CrystalCell.element1Basis element 1 element2Basis element 2 fmtstrFormat string. latticeStructure Crystal3DLattice.lattice_shiftLattice displacement vector. massTotal mass of atoms. n1Number of unit cells along Crystal3DLattice.a1.n2Number of unit cells along Crystal3DLattice.a2.r1Vector GrapheneMixin.n1\(\times\)Crystal3DLattice.a1.r2Vector GrapheneMixin.n2\(\times\)Crystal3DLattice.a2.scaling_matrixCrystalCell.scaling_matrix.structureAn alias to self.unit_cellStructure UnitCell.vdw_distanceVan der Waals distance. vdw_radiusVan der Waals radius Methods
clear()Clear list of StructureMixin.atoms.finalize()Finalize structure data by assigning unique ids and types to structure atoms. from_conventional_cell(**kwargs)classmethod from_primitive_cell(**kwargs)classmethod generate([finalize])Generate the full structure coordinates. generate_fname([armchair_edge_length, ...])Generate a filename string. make_supercell(scaling_matrix[, wrap_coords])Make supercell. rotate(**kwargs)Rotate crystal cell lattice, basis, and unit cell. save([fname])Save structure data. todict()Return dictof constructor parameters.transform_lattice(scaling_matrix[, ...])Transform structure lattice. translate(t[, fix_anchor_points])Translate crystal cell lattice, basis, and unit cell. write(*args, **kwargs)Write structure data to file. write_data(**kwargs)Write LAMMPS data file. write_dump(**kwargs)Write LAMMPS dump file. write_pdb(**kwargs)Write pdb file. write_xyz(**kwargs)Write xyz file. - armchair_edge_length (float, optional) –