UnrolledSWNTGenerator¶
-
class
sknano.generators.UnrolledSWNTGenerator(*args, autogen=True, finalize=True, **kwargs)[source] [edit on github][source]¶ Bases:
sknano.generators.NanoStructureGenerator,sknano.core.structures.UnrolledSWNTClass for generating unrolled nanotube structures.
New in version 0.2.23.
Parameters: - m (n,) – Chiral indices defining the nanotube chiral vector \(\mathbf{C}_{h} = n\mathbf{a}_{1} + m\mathbf{a}_{2} = (n, m)\).
- ny, nz (nx,) – Number of repeat unit cells in the \(x, y, z\) dimensions
- basis ({
list}, optional) –List of
strs of element symbols or atomic number of the two atom basis (default: [‘C’, ‘C’])New in version 0.3.10.
- element2 (element1,) –
Element symbol or atomic number of basis
Atom1 and 2Deprecated since version 0.3.10: Use
basisinstead - bond (float, optional) – \(\mathrm{a}_{\mathrm{CC}} =\) distance between nearest neighbor atoms. Must be in units of Angstroms.
- Ly, Lz (Lx,) –
Length of bundle in \(x, y, z\) dimensions in Angstroms. Overrides the \(n_x, n_y, n_z\) cell values.
Changed in version 0.4.0: Changed units from nanometers to Angstroms
- fix_Lz (bool, optional) – Generate the nanotube with length as close to the specified
\(L_z\) as possible. If
True, then non integer \(n_z\) cells are permitted. - autogen (bool, optional) – if
True, automatically callgenerate. - verbose (bool, optional) – if
True, show verbose output
Notes
The
UnrolledSWNTGeneratorclass generates graphene using the nanotube unit cell defined by the chiral vector \(\mathbf{C}_{h} = n\mathbf{a}_{1} + m\mathbf{a}_{2} = (n, m)\). If you want to generate graphene with an armchair or zigzag edge usinglengthandwidthparameters, see theGrapheneGeneratorclass.See also
Examples
First, load the
UnrolledSWNTGeneratorclass.>>> from sknano.generators import UnrolledSWNTGenerator
Now let’s generate an unrolled \(\mathbf{C}_{\mathrm{h}} = (10, 5)\) SWCNT unit cell.
>>> unrolled_swnt = UnrolledSWNTGenerator(10, 5) >>> unrolled_swnt.save()
The rendered structure looks like:
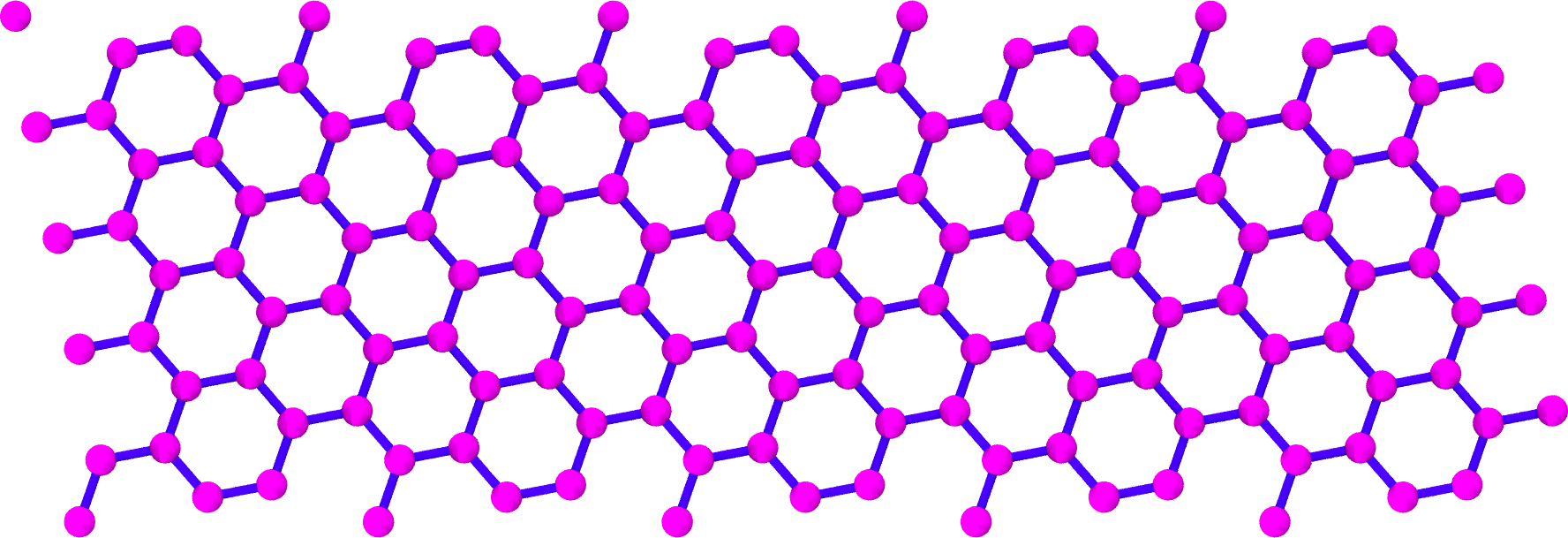
Attributes
ChSWNT circumference \(|\mathbf{C}_h|\) in Å Ch_vecSWNT chiral vector. LxAxis-aligned length along the x-axis in Angstroms.LyAxis-aligned length along the y-axis in Angstroms.LzSWNT length \(L_z = L_{\mathrm{tube}}\) in Angstroms. M\(M = np - nq\) NNumber of graphene hexagons in nanotube unit cell. NatomsN atoms. Natoms_per_layerNumber of atoms per layer. Natoms_per_tubeNumber of atoms in nanotube \(N_{\mathrm{atoms/tube}}\). Natoms_per_unit_cellNumber of atoms in nanotube unit cell. RSymmetry vector \(\mathbf{R} = (p, q)\). TLength of nanotube unit cell \(|\mathbf{T}|\) in Å. TvecSWNTtranslation vector.areaTotal area of graphene supercell. atomsStructure Atoms.basisNanoStructureBasebasis objects.chiral_angleChiral angle \(\theta_c\) in degrees. chiral_typeSWNTchiral type.crystal_cellStructure CrystalCell.d\(d=\gcd{(n, m)}\) dR\(d_R=\gcd{(2n + m, 2m + n)}\) dtNanotube diameter \(d_t = \frac{|\mathbf{C}_h|}{\pi}\) in Å. electronic_typeSWNT electronic type. element1Basis element 1 element2Basis element 2 fix_Lxboolindicating whetherUnrolledSWNTMixin.Lxis fixed or calculated.fix_Lzboolindicating whetherSWNTMixin.Lzis fixed or calculated.fmtstrFormat string. latticeStructure Crystal3DLattice.lattice_shiftLattice displacement vector. linear_mass_densityLinear mass density of nanotube in g/Å. mChiral index \(m\). massTotal mass of atoms. nChiral index \(n\). n1Number of unit cells along Crystal3DLattice.a1.n2Number of unit cells along Crystal3DLattice.a2.nlayersNumber of layers. nxNumber of unit cells along the \(x\)-axis. nyAn alias for UnrolledSWNTMixin.nlayers.nzNumber of nanotube unit cells along the \(z\)-axis. r1Vector GrapheneMixin.n1\(\times\)Crystal3DLattice.a1.r2Vector GrapheneMixin.n2\(\times\)Crystal3DLattice.a2.rtNanotube radius \(r_t = \frac{|\mathbf{C}_h|}{2\pi}\) in Å. scaling_matrixCrystalCell.scaling_matrix.structureAn alias to self.t1\(t_{1} = \frac{2m + n}{d_{R}}\) t2\(t_2 = -\frac{2n + m}{d_R}\) tube_lengthAlias for SWNT.Lztube_massAn alias for mass.unit_cellStructure UnitCell.unit_cell_massUnit cell mass in atomic mass units. unit_cell_symmetry_paramsTuple of SWNTunit cell symmetry parameters.vdw_distanceVan der Waals distance. vdw_radiusVan der Waals radius Methods
clear()Clear list of StructureMixin.atoms.finalize()Finalize structure data by assigning unique ids and types to structure atoms. generate([finalize])Generate structure data. generate_fname([n, m, nx, nz, fix_Lx, fix_Lz])generate_unit_cell()Generate the nanotube unit cell. make_supercell(scaling_matrix[, wrap_coords])Make supercell. rotate(**kwargs)Rotate crystal cell lattice, basis, and unit cell. save([fname, outpath, structure_format, ...])Save structure data. todict()Return dictof constructor parameters.transform_lattice(scaling_matrix[, ...])Transform structure lattice. translate(t[, fix_anchor_points])Translate crystal cell lattice, basis, and unit cell. write(*args, **kwargs)Write structure data to file. write_data(**kwargs)Write LAMMPS data file. write_dump(**kwargs)Write LAMMPS dump file. write_pdb(**kwargs)Write pdb file. write_xyz(**kwargs)Write xyz file. Methods Summary
generate([finalize])Generate structure data. generate_fname([n, m, nx, nz, fix_Lx, fix_Lz])save([fname, outpath, structure_format, ...])Save structure data. Methods Documentation
-
generate(finalize=True)[source] [edit on github][source]¶ Generate structure data.
-
classmethod
generate_fname(n=None, m=None, nx=None, nz=None, fix_Lx=False, fix_Lz=False, **kwargs)[source] [edit on github][source]¶
-
save(fname=None, outpath=None, structure_format=None, center_centroid=True, **kwargs)[source] [edit on github][source]¶ Save structure data.
See
savemethod for documentation.